характеристика относительной хим. активности молекул, атомов, ионов, радикалов. Для количеств. оценки Р. с. рассматривают реакционные серии, т. е. ряды однотипных р-ций, проводимых в одинаковых условиях, напр.: (стандартная р-ция)
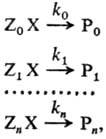
где Х-общая группа атомов, к-рая претерпевает изменения в данной р-ции (реакционный центр), Z0, Zl,...,
Типичные реакционные серии. Простейшая ситуация возникает при анализе изомерного состава продуктов р-ции. В р-ции электроф. замещения в ароматич. ряду в зависимости от заместителя R образуются те или иные изомеры, напр. при нитровании:

Электронодонорные заместители [R=СН 3, ОСН 3, N(CH3)2] стимулируют образование орто- и пара -продук-тов, а электроноакцепторные (R = СООН, SO3H, NO2)-мета -продуктов, причем в первом случае р-ция идет легче, чем с незамещенным бензолом (R = Н), а во втором-труднее. Эти закономерности наз. правилами ориентации в ароматическом ряду. При нуклеоф. замещении правила ориентации обращаются.
Стереохим. направленность перипиклич. р-ций определяется Вудворда-Хофмаиа правилами, напр.:
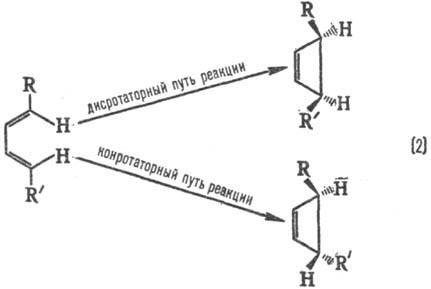
При дисротаторном пути р-ции заместители R и R' в продукте будут расположены по одну сторону плоскости цикла, при конротаторном пути-по разные стороны. Эксперимент показывает, что термич. циклизация производных бутадиена происходит по конротаторному пути, а фотохим. циклизация-по дисротаторному пути.
В примерах (1) и (2) нет необходимости в количеств. кинетич. измерениях, Р. с. определяется по относит. выходу изомеров. Пример широкой реакц. серии-р-ции радикального присоединения по двойной связи:

Р. с. характеризуется отношением константы скорости kк константе скорости k0 р-ции с этиленом (R, R' = Н) (см. табл.). Аналогичные кинетич. измерения сделаны для р-ций присоединения метильного радикала к ароматич. молекулам и для р-ций присоединения др. радикалов.

Квантовохимическая теория Р. с. Совр. теоретич. химия позволяет непосредственно рассчитать абс. константы скорости только для несложных хим. систем. В теории Р. с. качеств. закономерности м. б. выявлены для объектов любой сложности. При этом используют разл. подходы. При эмпирич. подходе классифицируют влияние заместителей по неск. типам (эффекты сопряжения, полярные, пространственные и др.) и применяют корреляционные соотношения. Традиц. квантовохим. подход основан на активированного комплекса теории; при этом предполагается, что для всех р-ций, составляющих реакц. серию (без пространств. и соль-ватац. эффектов), остается примерно постоянным пред-экспоненц. множитель Ав Аррениуса уравнении для константы скорости k = Aexp(-E./RT>)(R-газовая постоянная, Т-абс. т-ра). Поэтому характеристикой Р. с. служит энергия активации р-ции
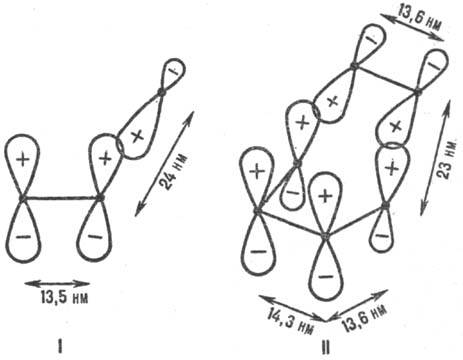
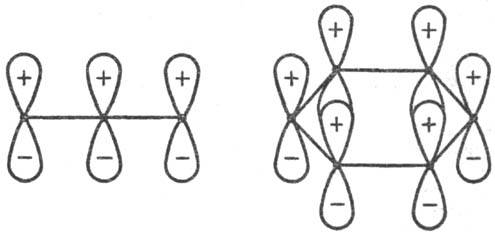
Рис. 1. Переходные состояния р-ций присоединения с участием p-электронных систем. I-радикальное присоединение (метальный радикал + этилен), II-р-ция Дильса-Альдера (этилен + бутадиен); внизу изображены соответствующие модельные p-сопряженные структуры. Показаны атомные 2pp -орбитали, асимметрия к-рых обусловлена изменением гибридизации вследствие взаимод. в переходном состоянии. Черные точки-ядра атомов С. Знаки " + " и " Ч" относятся к соответствующим волновым ф-циям. Орбитали перекрываются в фазе. Приведены межатомные расстояния в переходном состоянии.
Энергия стабилизации. Для относит. оценки
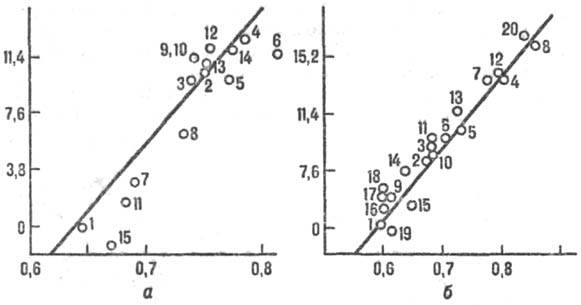
Рис. 2. Корреляция между реакц. способностью и энергией стабилизации переходного состояния, а-р-ции присоединения метального радикала СН Х3 к олефи-нам, цифры соответствуют номеру олефина в таблице. На оси ординат отложены величины 2,3 RТ lg k/k0 (кДж/моль), где k/k0 - отношение констант скорости рассматриваемой р-ции и р-ции с этиленом, R- газовая постоянная, Т-абс. т-ра. На оси абсцисс отложены энергии стабилизации в относит единицах, б-р-ция СН Х3 + ароматич. молекула. В скобках указано положение в молекуле субстрата, для к-рого приводится константа k. k0- константа скорости р-ции с бензолом. 1-бензол; 2-нафталин (1); 3-фенантрен (9); 4-антрацен (9); 5-пирен (1); 6-хризен (6); 7-бенз(а)антрацен (7); 8-нафтацен (5); 9-пиридин (2); 10-хино-лин (4); 11 -изохинолин (1); 12-акридин (9); 13-феназин (1); 14-бензонитрил (4); 15-ацетофенон (4); 16-фторбензол (4); 17-хлорбензол (4); 18-бромбензол (4); 19-анизол (4); 20-бензохинон.
Концепция граничных орбиталей. Оценки Р. с. особенно просты, если использовать возмущений теорию. В распространенном варианте теории возмущений энергия стабилизации представляется в виде суммы вкладов от взаимод. между мол. орбиталями реагентов. Наиб. вклад в сумму дают, как правило, взаимод. граничных орбиталей, т. е. высших заполненных электронами и низших незаполненных орбиталей; согласно К. Фукуи (1952), существенны только эти вклады (см. Граничных орбиталей теория). Концепцию граничных орбиталей часто применяют в качестве основы для обсуждения Р. с.
Альтернантные системы. Качеств. подход, не обязательно использующий теорию возмущений, сформулирован для класса сопряженных систем, наз. альтернант-ными. Они образованы из одинаковых атомов (обычно углерода) и не содержат нечетных циклов (см. Альтернантные углеводороды).Для таких систем в рамках Хюккеля метода можно без всяких вычислений выявить нек-рые общие закономерности. Так, введение полярного заместителя приводит к чередованию положит. и отрицат. изменений электронной плотности в сопряженной углеродной цепи относительно незамещенного углеводородного соед. (закон альтернирующей полярности, Ч. Коул-сон, Г. Лонге-Хиггинс, 1947). Этот вывод теории позволяет объяснить правила ориентации в ароматич. ряду (р-ция 1).
Для циклич. переходного состояния (активир. комплекса) существенно, каким образом замыкаются новые связи: в фазе или в противофазе, т. е. имеют ли атомные орбитали реагентов в области макс. перекрывания одинаковые или противоположные знаки (рис. 1 и 3). В первом случае взаимод. наз. связывающим, во втором-разрых-ляющим. В зависимости от числа атомных орбиталей в сопряженной системе активир. комплекса, электронного состояния реагирующей системы и характера вновь возникающих взаимодействий, энергия замыкания цикла м. б. как положительной, так и отрицательной, причем ее знак определяется без вычислений. В частности, при циклизации производных бутадиена (р-ция 2) в основном электронном состоянии переходное состояние стабилизируется замыканием связей посредством разрыхляющих взаимод. (перекрывание в противофазе, рис. 3), делая энергетически выгодным конротаторный путь термич. р-ции. В первом электронном возбужденном состоянии циклич. переходное состояние стабилизируется связывающим взаимод. (перекрывание в фазе), что соответствует дисротаторному пути. Эти общие положения позволяют предсказывать закономерности Р. с. согласованных электроциклич. р-ций (Р. Вуд-ворд, Р. Хофман, 1965).

Рис. 3. Переходные состояния р-ции циклизации производных бутадиена. I-конротаторный путь р-ции (орбитали вновь образующейся связи перекрываются в протпвофазе); II-дисротаторный путь р-ции (орбитали перекрываются в фазе). Обозначения те же, что и на рис. 1. Стрелки указывают области наилучшего перекрывания атомных орбиталей реакц. центра.
Индексы Р. с. - теоретич. величины, к-рые используют для характеристики Р. с. на простом модельном уровне и обычно рассчитывают квантовохим. методами. Один из индексов Р. с.-энергия стабилизации; др. индексы менее универсальны, часто они являются приближенными оценками энергии стабилизации применительно к конкретным типам р-ций. Так, в случае ионных р-ций с участием p-элект-ронных систем в качестве индексов Р. с. применяют p-электронные поляризуемости (для углеводородных субстратов) и p-электронные эффективные заряды атомов (для углеводородных субстратов с полярными заместителями или для гетероциклич. субстратов). В случае радикальных р-ций присоединения в качестве индексов Р. с. особенно подходящи энергии локализации, а также индексы своб. валентности. В частности, опытные константы скорости присоединения метильного радикала к олефинам и ароматич. молекулам (рис. 2) удовлетворительно коррелируют с энергиями локализации. Совр. развитие представлений об индексах Р. с. связано с т. наз. теорий функционала плотности, согласно к-рой для квантовомех. описания электронного строения мол. систем вместо волновой ф-ции используется ф-ция электронной плотности (Р. Парр, В. Янг, 1989).
Индексы Р. с. применимы в тех случаях, когда хим. перестройка затрагивает в осн. p-электронные системы реагентов. Если же наряду с p-электронной системой значительно перестраиваются и s-связи реагентов, применение p-электронного приближения мало эффективно. В подобных случаях, а также если в реагирующих системах вообще нельзя выделить систему p-электронов, изменения энергий активации р-ций, составляющих реакц. серию, оцениваются с помощью полуэмпирич. или неэмпирических методов квантовохим. вычислений в рамках разл. моделей р-ции, учитывающих все валентные электроны. В результате получают индексы Р. с., к-рые при подходящем выборе модели удовлетворительно коррелируют с кинетич. изменениями. Такое квантовохим. описание утрачивает качеств. наглядность p-электронной модели.
Соотношение Брёнстеда. Если простые квантовохим. методы неэффективны, Р. с. часто интерпретируют с помощью определяемых на опыте корреляций между логарифмами констант скорости i
(рис. 4). В предположении о постоянстве пред-экспоненц. множителя в ур-нии Аррениуса для

(В и А-радикалы или анионы; А
Химическая энциклопедия. — М.: Советская энциклопедия. Под ред. И. Л. Кнунянца. 1988.