DISTILLATION
La distillation est une des méthodes de séparation les plus importantes dont on dispose pour résoudre un mélange en ses divers constituants. Elle est fondée sur le fait qu’une phase vapeur surmontant un liquide bouillant et en équilibre avec ce dernier n’en a pas la même composition. En recueillant séparément, d’une part, le liquide et, d’autre part, la vapeur que l’on refroidit et condense, on obtient deux liquides différents appelés respectivement résidu et condensat ou distillat. On réalise ainsi une étape élémentaire de séparation dont la répétition pourra permettre d’obtenir les constituants du mélange initial.
Bien que le premier traité à ce sujet, œuvre du chimiste français Arnaud de Villeneuve, ne soit paru qu’aux environs de 1311, l’«art de la distillation» daterait de plus de trois mille ans, et l’on pense que les Perses l’auraient découvert pour fabriquer l’eau de rose. Rappelons aussi qu’Aristote proposait aux marins de distiller l’eau de mer pour produire de l’eau douce.
Les premiers véritables appareils à distiller furent conçus par les coptes d’Alexandrie et les chrétiens d’Égypte; ils comprenaient une cornue (cucurbite), un récipient de condensation des vapeurs (ambix) et un réceptacle des produits distillés (phiale). Le terme ambix a donné « alambic », qui désigne l’appareil entier de distillation.
Les médecins et pharmaciens améliorèrent les appareils et la technique. Ainsi, les apothicaires du XIIe siècle surent préparer des médicaments alcoolisés dont l’usage se généralisa rapidement, Irlandais et Écossais se signalant alors par leur particulière compétence dans la préparation de ces boissons.
Les producteurs d’alcool furent d’ailleurs pendant longtemps les experts en distillation, et c’est grâce à leurs travaux, complétés principalement, depuis cinquante ans, par les recherches des ingénieurs de l’industrie pétrolière, que la distillation occupe une place industrielle aussi importante parmi les opérations de séparation. Pour la souligner, il faut rappeler que la distillation est l’opération de base du raffinage des pétroles bruts.
1. Équilibre liquide-vapeur d’un mélange binaire
Définitions
On exprimera ici la composition des mélanges en fractions moléculaires (rapport entre le nombre de molécules du constituant considéré et le nombre total correspondant de molécules du mélange). On les représentera par x pour les liquides et y pour les vapeurs. Un mélange binaire sera parfaitement défini par la connaissance de la fraction moléculaire en un seul constituant (x A par exemple), l’autre s’obtenant immédiatement par différence à l’unité (x B = 1 漣 x A).
La volatilité d’un constituant A d’un mélange s’exprime par le rapport de ses concentrations respectivement en phase vapeur et en phase liquide, c’est-à-dire KA = y A/x A
La distillation dépend finalement des différences de volatilité entre les constituants de la charge à séparer; c’est afin de comparer les volatilités respectives que l’on a introduit la volatilité relative 見AB définie par le rapport des volatilités:

Les volatilités dépendent à la fois de la pression et de la température auxquelles sont soumises la phase vapeur et la phase liquide en équilibre ainsi que des interactions s’exerçant entre les molécules de ces milieux.
On conçoit que plus 見AB s’écartera de l’unité, plus aisée sera la séparation par distillation des corps A et B.
Si on veut préciser les définitions précédentes, on donne la composition des phases vapeur y et liquide x en équilibre à une température donnée et sous une certaine pression ainsi que les volatilités K et volatilités relatives 見 établies en choisissant convenablement un constituant de référence.
Considérons le cas simple d’un liquide pur à l’ébullition; il est surmonté d’une vapeur exerçant sur les parois du récipient une certaine pression, dite tension de vapeur, pression qui résulte des chocs exercés par les molécules à l’état gazeux contre le récipient. La tension de vapeur d’un corps dépend de la température et croît avec cette dernière (fig. 1).
Examinons maintenant un mélange binaire (constituants A et B) que l’on chauffe ou refroidit sous une pression donnée; à la différence du comportement du corps pur précédent, deux cas sont à étudier:
– le mélange liquide initial est chauffé, il commencera à bouillir à une température donnée unique qui est sa température d’ébullition commençante eb.
– le mélange gazeux initial est refroidi, il commencera à se condenser à une température donnée unique qui est sa température de condensation ou température de rosée r.
La température de rosée est supérieure à la température d’ébullition commençante. La figure 2 résume les remarques précédentes: ML représente le mélange liquide initial de composition x MA. Lorsque ce mélange est à sa température d’ébullition commençante, sous pression P0, il est représenté par I. Mv représente le mélange gazeux (teneur y MA), qui est représenté par J lorsqu’il atteint sa température de rosée, sous la pression P0.
Sous la pression P0 et à la température T, le mélange M donne une phase liquide L à sa température d’ébullition commençante et une phase vapeur V à sa température de rosée. Les phases étant en équilibre, ces deux températures sont égales. La volatilité relative 見AB sera d’autant plus grande que les points L et V seront distants.
Pour une autre température, il s’établirait, sous la même pression P0, un équilibre différent, et l’ensemble des points représentatifs respectivement des liquides et vapeurs aux différentes températures définit la courbe d’ébullition et celle de rosée. Ces deux courbes se rejoignent en deux points correspondant respectivement à chacun des corps purs à son point d’ébullition (TA et B) sous la pression considérée. À titre d’exemple, la figure 3 montre un tel fuseau relatif à l’équilibre d’un mélange d’hydrocarbures sous la pression atmosphérique.
Il est souvent utile d’exprimer la composition de la phase vapeur y en fonction de celle de la phase liquide x en équilibre; on obtient ainsi la courbe dite y . x (fig. 4). L’équation de cette courbe est, d’après la définition de la volatilité relative:

Solutions idéales
Si les interactions entre les molécules sont telles que l’une quelconque de celles-ci se comporte de la même manière aussi bien au sein du mélange liquide avec d’autres constituants que dans sa phase liquide pure, on dit que le mélange est une solution idéale; l’application des lois de Dalton et de Raoult permet alors d’établir les relations KA = PA/P et KB = PB/P, où PA et PB sont les tensions de vapeur, à la température considérée, respectivement des corps A et B purs et P la pression d’équilibre. Il en résulte que 見AB = PA/PB. L’équilibre sous pression modérée est alors calculable à partir de la connaissance des tensions de vapeur des constituants purs du mélange.
Le plus souvent, on ne peut assimiler le liquide à une solution idéale; on conserve alors la forme des expressions précédentes et l’on fait intervenir des termes correctifs, dits coefficients d’activité 塚, qui sont déterminés généralement par l’intermédiaire d’informations expérimentales. On obtient ainsi:

Pour être complet, il faudrait introduire des termes supplémentaires traduisant, si nécessaire, l’effet de la pression sur le comportement de la phase vapeur.
Formation d’un azéotrope
On voit, d’après la dernière relation qu’en raison de l’influence des coefficients d’activité il peut se faire que 見AB égale l’unité; il en résulte, d’après l’équation reliant y à x , que ces dernières valeurs sont alors égales. Le liquide et la vapeur ont la même composition, il n’y a plus de séparation; on dit qu’il s’est formé un azéotrope. Les courbes de rosée et d’ébullition présentent alors un point commun auquel correspond, le plus souvent, la température la plus basse de l’équilibre (fig. 5).
De même que la température d’ébullition d’un corps croît avec la pression, la température d’ébullition commençante d’un mélange ou sa température de rosée augmente lorsque la pression croît. Il faut de plus préciser que cela s’accompagne d’une diminution de l’écart entre ces deux dernières températures, c’est-à-dire que les deux courbes du fuseau sont d’autant plus proches que la pression est élevée. Cela revient à dire que l’augmentation de la pression, entraînant une diminution de la volatilité relative 見AB, est défavorable à la séparation.
2. Mise en œuvre de la distillation
On distingue deux façons d’effectuer une distillation:
– En continu: l’appareil est alimenté de façon permanente, tandis qu’on soutire, à débits et compositions fixes, un distillat et un résidu respectivement en tête et au fond de l’appareil. En tout point de l’appareil, concentration, température, pression et débits sont indépendants du temps; on dit que le régime est stationnaire. C’est le mode opératoire le plus couramment retenu par l’industrie, surtout pour le traitement de quantités importantes de produits.
– En discontinu: il s’agit d’une opération évoluant dans le temps. La charge est initialement disposée dans l’appareil où elle est chauffée; sa composition varie ainsi que celle du distillat. Il n’y a pas de soutirage de résidu. Cette technique est fréquemment employée au laboratoire, elle est aussi celle de l’artisanat (c’est ainsi que procèdent les «bouilleurs de cru»).
Dans ce texte, on ne s’intéressera qu’à la distillation effectuée en continu.
Examinons d’abord le cas simple des appareils à un seul étage de contact entre le liquide et la vapeur (distillation continue simple). La figure 6 présente le schéma de l’appareil utilisé pour distiller une charge liquide. L’opération s’effectue à pression et température constantes. Pour suivre le fonctionnement de cette installation, on admettra qu’elle est équivalente à un étage théorique , c’est-à-dire que la vapeur et le liquide quittant le séparateur sont en équilibre.
Supposons que l’on distille, avec cet équipement fonctionnant à la pression atmosphérique, un mélange équimoléculaire d’isobutène et d’isoprène. La figure 3 montre que, lorsque l’appareil fonctionne à 7,2 0C, le pourcentage vaporisé de la charge est infime et que les bulles de vapeur contiennent 81,5 p. 100 d’isobutène. On constate aussi que, à 21,5 0C, la charge est entièrement vaporisée. Les températures de distillation sont donc situées entre 7,2 0C et 21,5 0C.
Finalement, il est impossible de séparer le mélange en ses constituants purs au moyen de cet appareil. Par contre, il est possible de fractionner la charge en deux nouveaux mélanges respectivement enrichis en isobutène et isoprène. Afin d’améliorer la séparation du mélange, il vient immédiatement à l’idée d’effectuer une succession de traitement en un étage.
Agencement des étages théoriques
Cascade de distillations continues simples
On dispose de plusieurs appareils analogues au précédent, que l’on cherche à faire fonctionner en série (fig. 7 a). La charge F, de composition x 0, donne, dans l’étage 1 fonctionnant à la température 1, un liquide L1 et une vapeur V1. L1 est chauffé et partiellement vaporisé à la température 2 dans l’étage 2, ce qui donne le liquide L2 qui, dans l’appareil suivant, donne L3. De même, la vapeur V1 est refroidie et partiellement condensée à la température T 2 dans l’étage 2 . La nouvelle vapeur produite V 2 donne à son tour dans l’élément suivant la vapeur finale V 3.
L’évolution des phases au cours d’un tel procédé est donnée par la figure 7 b. On constate que, d’une part, les équilibres n’ont pas lieu à la même température; en effet:
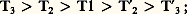
d’autre part, les liquides L1, L2, L3, en se réchauffant, s’enrichissent en l’élément le plus lourd de la charge; enfin, les vapeurs V1, V 2, V 3, en se refroidissant, s’enrichissent en l’élément le plus léger du mélange traité. Par conséquent, si l’on dispose d’un grand nombre d’étages situés de part et d’autre de l’étage d’alimentation, on peut séparer le mélange en chacun de ses constituants presque purs. Il faut remarquer cependant que le rendement de cette opération n’est guère élevé, car seuls nous intéressent finalement la vapeur V 3 et le liquide L3, alors que les divers produits intermédiaires formés (liquides et vapeurs) doivent être rejetés. Afin de remédier à cette perte de rendement, on utilise, en réalité, un agencement des étages différent et réalisant un contre-courant entre les phases.
Cascade d’étage fonctionnant à contre-courant
La technique précédente montre tout l’intérêt qu’il y a à vaporiser ou condenser partiellement les effluents de chaque séparateur de distillation continue simple.
La rectification réalise ces vaporisations et condensations successives en faisant s’écouler à contre-courant une phase vapeur et une phase liquide. La vapeur chauffe le liquide dont les éléments légers se vaporisent; le liquide refroidit la vapeur dont les éléments lourds se condensent. Le transfert de chaleur s’accompagne donc d’un transfert de matière entre les phases.
Le problème pratique consiste à réaliser le contre-courant liquide-vapeur. Dans ce but, on effectue, d’une part, un recyclage de vapeur condensée au sommet de l’appareil et, d’autre part, une vaporisation partielle du liquide en bas de la colonne de distillation (fig. 8). L’alimentation, préalablement chauffée, se vaporise en partie dans sa zone d’admission dans la colonne. La vapeur produite monte dans la tour, traverse le condenseur où elle se liquéfie par refroidissement; une partie du liquide obtenu est soutirée (c’est le distillat), l’autre est réintroduite dans l’appareil (c’est le reflux liquide qui descend en croisant à contre-courant la vapeur); le reflux est particulièrement riche en élément volatil, ce qui provoque un enrichissement de la vapeur en ce constituant léger. Puis le liquide descend dans la colonne, il est partiellement vaporisé dans le rebouilleur; la vapeur émise monte dans l’appareil à contre-courant du liquide et en chassant de ce dernier ses constituants légers. Le résidu liquide est soutiré au fond de la colonne.
La section supérieure de la tour à distiller (au-dessus de l’introduction de la charge) est la zone d’enrichissement de la vapeur en élément le plus volatil; elle a pour but d’augmenter la pureté de l’élément léger constituant principalement le distillat; la section inférieure sert à épuiser le liquide en ce constituant volatil, c’est-à-dire à augmenter son taux de récupération.
Dans une telle colonne, on fournit de la chaleur aux produits par le rebouilleur (dans le bas de l’appareil) et on l’élimine au sommet par l’intermédiaire du condenseur.
Appareillage
Les appareils ont tous pour but de réaliser le meilleur échange de matière entre le liquide et la vapeur qui se croisent. Ils doivent donc être équipés de dispositifs internes qui, d’une part, favorisent la dispersion de la vapeur dans le liquide et plus particulièrement provoquent la plus grande surface d’aire interfaciale, et, d’autre part, permettent la séparation du liquide et de la vapeur en contact, afin d’en faciliter l’écoulement global à contre-courant. Les appareils les plus couramment employés sont équipés de plateaux sur lesquels s’effectue le contact entre les phases – et, par conséquent, l’échange de matière – et entre lesquels la vapeur et le liquide s’écoulent à contre-courant sans être, le plus souvent, en contact (fig. 9). Différents types de plateaux peuvent être employés; les plateaux à calottes, jusqu’à maintenant les plus utilisés, sont progressivement remplacés par d’autres dispositifs (plateaux à clapets, par exemple) de construction moins coûteuse. La figure 9 permet de suivre le fonctionnement d’un plateau. Le liquide est admis sur le plateau par le déversoir du plateau supérieur et s’en échappe par son propre déversoir après avoir circulé entre les calottes. La vapeur pénètre dans la cheminée de la calotte, s’écoule par les fentes situées à sa base et traverse, sous forme de bulles, la couche de liquide surmontant les fentes.
Si le diamètre de la tour à distiller devient important, on aura recours alternativement à un déversoir central et à deux déversoirs latéraux.
Le plus souvent, les appareils de laboratoire ou les unités industrielles de faible capacité ne comportent pas de plateaux mais sont équipés d’éléments de remplissage qui ont pour but de faciliter le contact entre la vapeur et le liquide. La disposition d’une colonne à remplissage est analogue à celle d’une colonne à plateaux, ces derniers étant remplacés par des corps solides remplissant tout l’espace de la section correspondante. Les éléments de remplissage peuvent être constitués de matériaux les plus divers: porcelaine, verre, acier, plastiques... Les plus couramment employés sont les anneaux Rashig (cylindres creux, de hauteur égale à leur diamètre) et les anneaux Pall.
Calcul d’une colonne
Le calcul d’une tour à distiller exige la résolution de plusieurs problèmes. Il faut en particulier:
– chiffrer la difficulté de la séparation à réaliser (c’est-à-dire évaluer le nombre d’étages théoriques nécessaires pour effectuer la séparation) ainsi que le reflux et les quantités de chaleur à fournir au rebouilleur et à éliminer au condenseur; cela ne dépend que des débits des produits circulant dans l’appareil (équations de bilan) et des données d’équilibre relatives au mélange traité (ce qui implique qu’il faut avoir fait choix, au préalable d’une pression de fonctionnement);
– chiffrer l’efficacité de l’appareil, c’est-à-dire déterminer le nombre de plateaux réels (ou la hauteur de garnissage) équivalant au nombre d’étages théoriques trouvé précédemment; cela fait appel à la cinétique des transferts de matière et de chaleur, dont l’importance dépend des fluides en présence, et du régime d’écoulement réalisé à l’endroit où s’effectue le transfert (calotte des plateaux en ce qui concerne le transfert de matière).
Choix de la pression de fonctionnement
La distillation est une opération qui peut s’effectuer sous des pressions extrêmement différentes. Le choix de la pression est ordinairement fixé afin de pouvoir condenser, tout au moins partiellement, la vapeur quittant le sommet de la colonne par échange de chaleur avec un fluide peu coûteux (le plus souvent de l’eau). On cherche aussi à vaporiser une partie du liquide dans le rebouilleur par échange de chaleur avec un produit convenable (vapeur d’eau si possible). Quand ces deux conditions ne peuvent être réalisées simultanément, il pourra être nécessaire soit d’effectuer la condensation à basse température en faisant appel à une machine frigorifique coûteuse, soit d’effectuer la vaporisation par chauffage direct dans un four.
La distillation sous pression sera employée lorsque la charge traitée est très volatile. L’augmentation de la pression provoque l’augmentation de la température de rosée, la vapeur peut alors être condensée par de l’eau. Elle ne pourra pas être pratiquée, d’une part, si les produits ne sont pas stables thermiquement, et, d’autre part, si le liquide de fond de tour atteint ou dépasse sa température critique; en effet, dans ce dernier cas, il ne peut exister une vapeur et un liquide en équilibre, on ne trouve alors qu’une phase unique, dite supercritique. La réalisation du contre-courant nécessaire à la distillation devient évidemment impossible.
On fera appel à la distillation sous pression inférieure à la pression atmosphérique lorsqu’on aura à distiller un mélange contenant soit des produits fort peu volatils, soit des produits instables thermiquement.
3. Distillations particulières
La distillation ordinaire ne peut être employée pour séparer un mélange binaire en ses constituants si ces derniers forment un azéotrope à la pression considérée. En effet, si le mélange a la composition de l’azéotrope, la distillation n’amène aucune séparation: liquide et vapeur ont la même composition; si le mélange diffère de l’azéotrope, la distillation – même effectuée avec un appareil extrêmement efficace – ne fournira que l’un des constituants purs et l’azéotrope.
La séparation correcte et économique des mélanges de constituants de volatilités voisines ne pourra non plus s’effectuer par distillation simple. On a donc cherché à modifier les conditions opératoires de la distillation afin de l’adapter à la résolution des problèmes précédents. Dans ce but, on modifie les équilibres liquide-vapeur en incorporant un solvant dans le mélange traité. On effectue ainsi soit une distillation extractive, soit une distillation azéotropique. Ces deux opérations nécessitent évidemment un traitement complémentaire afin de récupérer le solvant de séparation.
Distillation extractive
Comme cela a été déjà indiqué, la volatilité relative des divers constituants dépend des coefficients d’activité de chacun de ces constituants selon 見12 = 塚1P1/ 塚2P2.
L’addition d’un solvant dans le milieu bouleverse les interactions moléculaires, ce qui modifie le rapport des coefficients d’activité 塚1/ 塚2 et, par suite, la volatilité relative. Cette action est d’autant plus importante que les constituants du mélange à traiter sont plus «chimiquement» différents.
Le solvant est injecté au sommet de la tour à distiller et s’écoule en restant en phase liquide dans l’appareil; il sort du rebouilleur mélangé à certains constituants de la charge. Les autres éléments de l’alimentation sont récupérés dans le distillat (fig. 10). Une telle opération exige que l’on dispose d’un solvant de sélectivité convenable – c’est-à-dire modifiant favorablement les volatilités – et suffisamment peu volatil pour qu’il reste dans le liquide où s’exerce son action.
C’est par une distillation de ce type qu’il est possible d’extraire le butadiène contenu dans certaines coupes pétrolières qui sont des mélanges d’hydrocarbures à quatre atomes de carbone. Parmi les solvants utilisables pour cette opération, il faut citer l’acétonitrile ou la diméthylformamide.
Distillation azéotropique
Le solvant, introduit dans la charge forme un azéotrope avec un ou plusieurs de ses constituants. On recueille donc, dans le distillat, le solvant mélangé à quelques constituants de l’alimentation; les éléments complémentaires de cette dernière se retrouvent dans le résidu. À titre d’exemple, nous rappellerons que la déshydratation de l’alcool s’effectue par distillation azéotropique faisant appel au benzène comme solvant (fig. 11).
distillation [ distilasjɔ̃ ] n. f.
• 1372; bas lat. distillatio
♦ Procédé de purification (d'un liquide peu volatil, d'un corps solide : bois, houille) par ébullition suivie d'une condensation de la vapeur dans un autre récipient. Distillation dans un alambic. La distillation des hydrocarbures, de la houille. Colonne à distillation. Distillation fractionnée : séparation des constituants d'un mélange liquide fondée sur la différence de leurs températures d'ébullition. ⇒ rectification; azéotrope. Sous-produits de la distillation fractionnée du pétrole. Extraire, purifier par distillation. Distillation des moûts, des mélasses, des fruits, des grains, qui donne de l'eau-de-vie. Distillation de plantes aromatiques (⇒ essence) .
● distillation nom féminin (latin médiéval distillatio, -onis) Action de distiller ; procédé de séparation des constituants d'un mélange par ébullition. Traitement par la chaleur de certains produits agricoles en vue d'obtenir de l'alcool éthylique. ● distillation (expressions) nom féminin (latin médiéval distillatio, -onis) Distillation sèche, chauffage en atmosphère inerte de substances organiques (bois, charbons, schistes, huiles minérales résiduelles) pour en provoquer la décomposition chimique.
distillation
n. f. Opération qui consiste à faire passer un mélange liquide à l'état de vapeur, de façon à séparer ses divers constituants. Le principe de la distillation repose sur le fait que des substances mélangées ont, à une température donnée, des pressions de vapeur différentes.
— Distillation des vins, des fruits, des moûts, etc., qui donne les liqueurs alcooliques.
— Distillation fractionnée, pour séparer des liquides inégalement volatils.
⇒DISTILLATION, subst. fém.
CHIM. Opération par laquelle on sépare, au moyen du feu et dans des appareils fermés (cornues, alambics), des substances composées pour en recueillir les parties volatiles. Distillation de l'eau, des liqueurs; distillation lente, subtile; art de la distillation; soumettre à la distillation. La distillation de l'eau de mer (GOLDSCHMIDT, Avent. atom., 1962, p. 269) :
• 1. C'est encore dans le dix-septième siècle que l'usage de l'eau-de-vie commença à se répandre. La distillation, dont la première idée avait été apportée par les croisés, était jusque là demeurée un arcane qui n'était connu que d'un petit nombre d'adeptes.
BRILLAT-SAVARIN, Physiol. du goût, 1825, p. 275.
— Au fig. Ce qui s'écoule lentement; ce qui s'exprime de manière subtile ou raffinée. La distillation des siècles. La distillation, que nous avons créée, amènera des analyses plus intimes encore (RENAN, Drames philos., Eau jouvence, 1881, IV, 1, p. 486) :
• 2. Mais il existe un autre amour qui a besoin de la présence continue, de la personne tout entière, et qui s'approfondit par la durée (...) Il n'est pas cristallisation, mais distillation, lente élaboration d'essences précieuses.
CHARDONNE, Attachements, 1943, p. 24.
Prononc. et Orth. :[ ]. Ds Ac. 1694-1932. Cf. distiller. Étymol. et Hist. 1. a) 1372 [éd. 1522] distillation de l'urine (CORBICHON, Des eaux artificielles, à la suite des Propriétés des choses ds DELB. Notes [ms. Sorbonne]); b) 1677, 15 oct. fig. des distillations et des distinctions métaphysiques (Mme DE SÉVIGNÉ, Lettre à Madame de Grignan, éd. Ad. Regnier, t. 5, p. 366); c) 1694 « la chose distillée » distillations précieuses (Ac.); 2. XVe s. « action de ce qui tombe, ce qui tombe goutte à goutte » (CHASTELLAIN, Chron., éd. Kervyn de Lettenhove, V, 299). Empr. au lat. médiév. distillatio « distillation » (1267 ds LATHAM), dér. de distillare, v. distiller. Fréq. abs. littér. :27.
]. Ds Ac. 1694-1932. Cf. distiller. Étymol. et Hist. 1. a) 1372 [éd. 1522] distillation de l'urine (CORBICHON, Des eaux artificielles, à la suite des Propriétés des choses ds DELB. Notes [ms. Sorbonne]); b) 1677, 15 oct. fig. des distillations et des distinctions métaphysiques (Mme DE SÉVIGNÉ, Lettre à Madame de Grignan, éd. Ad. Regnier, t. 5, p. 366); c) 1694 « la chose distillée » distillations précieuses (Ac.); 2. XVe s. « action de ce qui tombe, ce qui tombe goutte à goutte » (CHASTELLAIN, Chron., éd. Kervyn de Lettenhove, V, 299). Empr. au lat. médiév. distillatio « distillation » (1267 ds LATHAM), dér. de distillare, v. distiller. Fréq. abs. littér. :27.
]. Ds Ac. 1694-1932. Cf. distiller. Étymol. et Hist. 1. a) 1372 [éd. 1522] distillation de l'urine (CORBICHON, Des eaux artificielles, à la suite des Propriétés des choses ds DELB. Notes [ms. Sorbonne]); b) 1677, 15 oct. fig. des distillations et des distinctions métaphysiques (Mme DE SÉVIGNÉ, Lettre à Madame de Grignan, éd. Ad. Regnier, t. 5, p. 366); c) 1694 « la chose distillée » distillations précieuses (Ac.); 2. XVe s. « action de ce qui tombe, ce qui tombe goutte à goutte » (CHASTELLAIN, Chron., éd. Kervyn de Lettenhove, V, 299). Empr. au lat. médiév. distillatio « distillation » (1267 ds LATHAM), dér. de distillare, v. distiller. Fréq. abs. littér. :27.
distillation [distilɑsjɔ̃] n. f.
ÉTYM. 1372; bas lat. distillatio « écoulement », du supin de distillare. → Distiller.
❖
1 Techn. et cour. (sauf certains emplois). Procédé qui consiste à séparer les constituants d'un mélange, d'une solution liquide, en utilisant les variations des températures d'équilibre liquide-vapeur selon la composition. || La distillation d'un mélange dans un alambic. || Les substances volatiles que libère la distillation sont recueillies par condensation (produits de distillation), sous forme de vapeur (⇒ Hydrolat) ou de gaz. || La distillation sèche s'opère sans addition d'eau. || Distillation fractionnée : séparation d'un mélange de plusieurs liquides à points d'ébullition différents, recueillis successivement. ⇒ Cohobation, rectification. || Extraire, purifier par distillation. || Produit que donne la première chauffe dans la distillation (⇒ Flegme); produit de tête, de cœur, de queue. || Distillation qui débarrasse l'eau de ses sels. || Distillation du bois. ⇒ Créosote, goudron. — Gaz d'éclairage, charbon de cornue, coke, goudrons produits dans la distillation de la houille. || Distillation des pétroles, des térébenthines (⇒ Raffinage). — Cour. || Distillation des vins, des cidres, des marcs, des lies; des moûts de betterave, des mélasses de canne à sucre; des fruits, des grains, des pommes de terre et des topinambours… ⇒ Alcool, eau-de-vie. || Résidu de la distillation des grains (⇒ Drèche), des liqueurs alcooliques (⇒ Vinasse). || Distillation de plantes aromatiques (⇒ Essence).
0 Si (…) on soumet à la distillation un mélange de plusieurs corps volatils, ces corps distillent dans l'ordre de leurs points d'ébullition réfrigérant. Toutefois, la séparation obtenue n'est pas rigoureuse, car les corps les moins volatils, bien avant même que leur point d'ébullition n'ait été atteint, peuvent être entraînés par les vapeurs des corps les plus volatils.
Omnium agricole, Distillation.
➪ tableau Vocabulaire de la chimie.
REM. Dans la langue courante, le mot ne s'emploie guère qu'en parlant de la distillation alcoolique, et aussi des parfums.
2 Fig. et littér. Ce qui s'exprime lentement, de manière subtile (→ les valeurs fig. de Distiller, I., 1., et I., 4.).
Encyclopédie Universelle. 2012.